新藥的0→1:淺談臨床試驗

文/王鳳宇
最近吵得沸沸揚揚的新冠肺炎疫苗,使得臨床試驗的相關議題再度浮上檯面。臨床試驗跟新藥的研發息息相關,政府機關與藥廠要如何確定新研發出來的藥物有顯著的療效,而且沒有嚴重的副作用呢?「臨床試驗」在這中間架起了重要的橋梁。
這篇文章主要針對臨床試驗作概述。接下來將一一介紹如何從實驗室中的發現,一步步通過一至三期的臨床試驗,最後被美國食品藥物管理局(FDA)核准上架及快速通過試驗的特殊案例。
一、 臨床試驗分期
首先,讓我們以一張概念圖做為開場(圖一)。這張圖總結了一款新藥從臨床前的藥物開發、臨床前測試、一至三期試驗、第四期試驗(上市後監測)所花費的時間與受試者的數目。從圖中可以看出一款藥物從無到有是多麼的困難,需要多少受試者,更別說需要燒掉多少錢。

藥物開發(Drug Discovery)
這個階段可說是新藥開發的起點。全球各地的頂尖實驗室日以繼夜研究著各式各樣的致病機轉,為的就是點燃一個治療某種疾病的小希望。在這個階段,可能有數千甚至數萬種的候選藥物(如:抗體、小分子化合物、疫苗)。在經過無數嚴謹的實驗及分析後,篩選出幾百種候選藥物進到下個階段。
臨床前試驗(Preclinical Test)
前一個階段中的實驗畢竟不是在活體中進行,實驗時的條件也與真實生理環境不同。因此在這個階段中,科學家會利用模式動物(如:小鼠、大鼠)進行候選藥物的測試。基本上能通過這個階段的候選藥物,便能向FDA申請進入臨床試驗(Investigational New Drug Application, IND),也就是開始以人體進行試驗。
向FDA申請進行人體試驗後,藥廠便會委託(贊助)醫院協助試驗的進行,因此通常臨床試驗的主持人是專科醫生。
第一期試驗(安全性測試)
由於這些候選藥物從來沒有被用在人身上,醫生也不清楚究竟多少劑量的藥物會讓人體產生不良反應(副作用)。因此臨床試驗的第一期便是釐清:
(1) 人體能承受最大劑量(maximum tolerated dose, MLD, 註一)
(2) 動物與人體中藥物動力學參數差異(藥物吸收、半衰期、分布等),換言之就是看這款藥物在人體中會如何反應。
在第一期試驗中除了觀察藥物動力學參數外,還會針對生體可用率(bioavailability)、生體相等性(bioequivalent)等進行探討,此文略。
第一期試驗中通常會招募數位至數十位健康受試者進行安全性測試。但要注意的是,有些特殊的藥物(如:抗體,因其會引起免疫反應)會直接招募病患進行第一期試驗,因為若在健康人身上測試,將難以評估藥物風險及效益。
第二期試驗(療效驗證)
第二期試驗主要進行「概念性驗證(Proof of Concept)」,以驗證該藥物是否具有實際療效。此階段中主要探討:
(1)療效及藥物不良反應(adverse drug reactions, ADRs)。
(2)確立藥物最佳劑量、給藥細節(如:間隔時間、給藥次數),為第三期試驗做準備。
因第二期試驗開始評估療效及潛在風險,試驗招募病患會增加至幾百人,且第二期試驗常被分為IIa與IIb期。IIa期用以評估療效;IIb期用以決定最佳劑量。
第三期試驗(療效比較)
這個階段是新藥在被批准上市前的最後一道關卡,試驗規範及試驗設計皆極為嚴格。第三期試驗的設計有幾個關鍵字:
大型、隨機(註二)、對照
此試驗通常為跨國、多個醫學中心協同執行(考量到人種因素),收樣人數多為幾千至幾萬人。此時,試驗中會加入對照組(給予安慰劑或同類型藥物)。此目的在於評估該新藥療效是否大於等於同類型藥物。若療效低於現有藥物,讓該新藥上市也無太大的意義。試驗同時,醫療人員也會詳細記錄所有受試者身上發生的不良反應,若不良反應太嚴重(如:休克、死亡)或太常發生(如:發生率為70%),也會影響到試驗的通過與否。
此外,因為第三期試驗會將新藥與既有藥物、安慰劑進行比較,為避免受試者或醫師的主觀因素影響結果判斷,此試驗多採取盲化分析(註三),而試驗最後揭露實驗結果的過程就是各位常聽到的「解盲(unblind)」。
你可能會好奇,既然第三期試驗如此的嚴格、規模如此龐大,那麼新藥如何通過第三期試驗,被FDA核准上市呢?
事實上,每一期試驗都有所謂的試驗終點(endpoints)。試驗主持人可以決定主要或次要的試驗終點(primary/secondary endpoints),並以此作為該新藥是否能通過試驗上市的關鍵。以癌症治療藥物為例,常見的試驗終點列舉如下(各指標細節就不多贅述,有興趣可自行上網搜尋):
與病患存活有關
整體存活率(overall survival, OS):患者從試驗開始直至任何原因造成死亡之時間。
疾病無惡化存活期(progression-free survival, PFS):患者從試驗開始直到疾病惡化或死亡之時間。
與療效有關
客觀反應率(overall response rate, ORR):以腫瘤大小做為評估療效的指標;腫瘤大小的測量以電腦斷層掃描(CT)為準。其中又細分為完全緩解(complete response, CR)、部分緩解(partial response, PR)、疾病穩定(stable disease, SD)等。
當上述提到的終點有達標後,恭喜!藥廠可以將試驗的相關文件與紀錄送交FDA申請新藥上市程序(New Drug Application, NDA),進行新藥查驗登記。FDA審核通過後核發藥證,該藥物即可上市銷售。
第四期試驗(上市後監測)
你可能會有些疑惑,既然新藥都上架販賣了,那第四期試驗在幹嘛?
理由很簡單。因為在一到三期試驗中招募的受試者必須滿足特定的規範(inclusion/exclusion criteria)(如:65歲以上老年人或女性不得懷孕等)。然而,新藥一旦上市,接受該藥物的族群將不受任何限制,因此為了確保上市後該藥物依然沒有嚴重的不良反應,有時會進行第四期試驗(沒錯!第四期試驗為非強制性)。有興趣的讀者可以上網搜尋「沛麗婷」。
講了這麼多,那臨床試驗的成功率與花費資金究竟有多少呢?根據工研院IEK產業情報網協助提供的資料,2010年《Nature Review Drug Discovery》期刊曾統計過(摘自:關鍵評論網),如下圖示
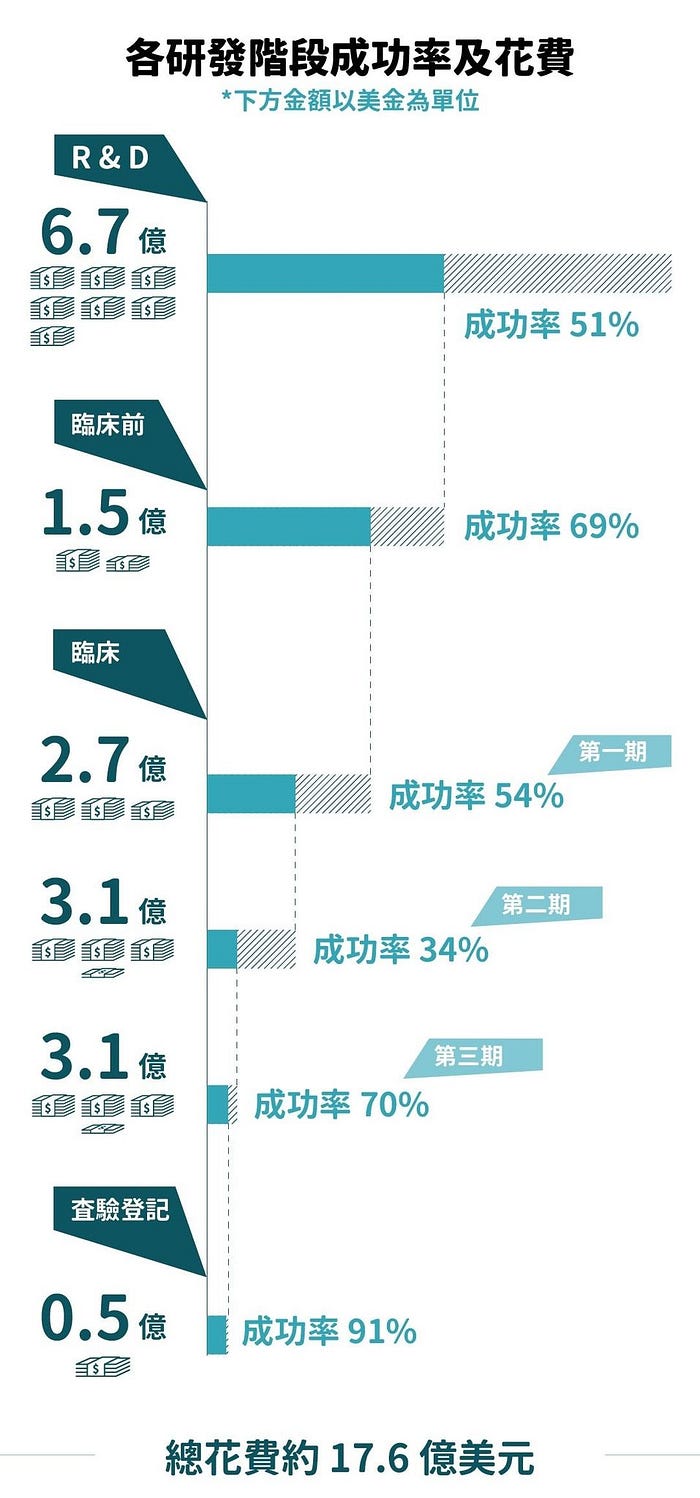
除了平均總花費約17.6億美元(折合台幣:528億元)外,若將成功的機率算一算,平均每100件試驗,約只有4件試驗能成功上市,可見一個新藥從無到有是多麼困難!
二、 通過臨床試驗的特殊方式
FDA為了因應特殊狀況而建立了一些藥物快速上架的方式,像是快速通道(fast track)、優先審查(priority review)、加速核准(accelerated approval)等,這邊我簡單介紹其中兩種方式。
加速核准(Accelerated Approval)
一款新藥通過三期試驗後會向FDA提出新藥審查。平均新藥審查程序約花費將近一年的時間。但許多重大疾病的病程變化快速,病患可能等不了這麼久,如:惡性腫瘤;或是治療方法非常稀少,如:罕見疾病。若遇到這種情形,這些藥物在試驗時可以採用替代指標做為試驗終點(endpoint),且FDA可以使用加速審查作業,使這些藥物較快問世,造福病人。
但畢竟這些藥物的上架速度較快,因此FDA也會監測該藥物上市後的具體療效及安全性,若發現效果不如預期或是不良反應過多,FDA會將該藥物下架。
緊急使用授權(Emergency Use Authorization, EUA)
因為新冠肺炎的關係,各位最近可能常在電視上看名嘴討論EUA。究竟EUA是什麼?
緊急使用授權(EUA)賦予FDA權力在緊急狀況時,緊急授權未核准使用的藥品或醫療器材進行疾病的診斷、治療及預防,這些緊急狀況包含了國防(恐怖攻擊)及公衛(嚴重傳染病)。舉例來說,除了這次的新冠肺炎外,像是2009年的H1N1流感病毒檢驗試劑、2013年的MERS-CoV、2014年的伊波拉病毒以及2016年的茲卡病毒(ZIKA),FDA都曾使用過EUA。
前面有提到,臨床試驗耗時又耗財。尤其是第三期試驗,試驗時程動輒三年起跳。那這些EUA授權的藥物或疫苗有沒有通過臨床試驗呢?在FDA的官網中有提到,這些藥物及疫苗皆須通過第一、二期臨床試驗,確認施打劑量且安全無虞後,進到第三期試驗。當第三期試驗進行到一半時,試驗方若有足夠的數據證明該藥物或疫苗的效用,便可以將數據提交至FDA申請EUA。若EUA通過了,則該藥物或疫苗便可以在三期試驗未終止前先行用來治療病患或做疾病的預防。
註:
一:多採用3+3試驗(3+3 dose escalation study),有興趣的讀者可以用這個關鍵字進行搜尋。
二:隨機指的是受試者被電腦隨機分配到對照組或實驗組,而非由試驗主持人或相關人士進行分配。
三:盲測又可分為單盲(single-blind)、雙盲(double-blind)、三盲(triple-blind)、甚至四盲(quadraple-blind)。單盲指的是只有研究者知道特定受試者被分至實驗組還是對照組。雙盲指的則是研究者與受試者雙方皆不知道。
來源:
1. https://clinicaltrials.gov/
2. https://www.thenewslens.com/article/95507
3.https://www3.cde.org.tw/Content/ebook/FlippingBook%20Files/RMV121/RMV121all/3/#zoom=z
